
2020년 의약품 제조.품질관리 기준 주요 개선사항 안내
담당부서 | 의약품품질과2020-12-02





2020년 의약품 제조·품질관리 기준 주요 개선사항 안내
「의약품 GMP 제도 개선사항 관련 질의·답변집」 발간
□ 식품의약품안전처(처장 김강립)는 올해 추진한 의약품 제조 및 품질관리기준(GMP)에 관한 국제조화 내용과 코로나19 확산에 따른 평가 방법 개편사항 등을 종합한 안내서를 발간합니다.
※ 의약품 제조 및 품질관리기준(GMP) : 품질이 보증된 의약품을 제조하기 위한 전 공정에 걸친 제조 및 품질관리에 관한 조직적이고 체계적인 규정
○ 이번 안내서는 올해 2월부터 10월까지 시행한 주요 제도 개선사항을 제약 현장에서 원활하게 적용할 수 있도록 마련하였으며, 업계의 이해도를 높이기 위해 그간의 주요 질의·답변 내용을 추가하였습니다.
○ 주요 내용은 ▲동일한 제조공정으로 위탁생산하는 전문의약품에 대한 자료제출 요건 강화 ▲전주기 관리를 위한 ‘의약품 품질시스템’ 도입 ▲의약품 허가신청 자료 및 품질관리의 신뢰성 향상을 위한 ‘의약품 제조·수입업체 데이터 완전성 평가지침’ 마련 ▲코로나19 확산 및 장기화에 따라 단계적으로 시행하는 수입의약품 GMP 평가 개편사항 등입니다.
□ 식약처는 앞으로도 국내 제약업계에서 국제적 수준의 제조 및 품질관리기준(GMP)을 도입·활용하도록 하여 보다 우수한 품질의 의약품을 국민께 공급하기 위해 노력하겠다고 밝혔습니다.
○ 자세한 내용은 식약처 대표누리집(mfds.go.kr) → 정책정보 → 의약품정책정보 → 제조품질관리기준(GMP)에서 확인할 수 있습니다.
<첨부> 2020년 의약품 GMP 제도 주요 개선사항
<첨부> 2020년 의약품 GMP 제도 주요 개선사항
? 의약품 등의 안전에 관한 규칙 (총리령) 개정
❍ (배경) 전공정 위탁제조 의약품의 허가(신고) 신청 자료를 강화하여 시장 건전성 및 품질확보
❍ (개정사항) 전공정 위탁제조 품목(전문의약품) GMP 평가 자료제출 의무화(‘20.10.14)
현 행
개 정 안 (개선 후, ‘20.10.14 개정완료)
제4조(제조판매ㆍ수입 품목의 허가 신청)
② 제1항에도 불구하고 다음 각 호의 어느 하나에 해당하는 경우에는 각 호의 구분에 따른 자료를 제출하지 아니할 수 있다.
3. 제조판매 품목허가를 신청하는 품목을 그 품목과 동일한 품목에 대하여 제조판매 품목허가를 받거나 신고를 한 자의 제조소에 해당 제조소의 품목과 모든 제조공정을 동일하게 제조하도록 위탁하는 경우: 제1항제6호에 해당하는 자료
제4조(제조판매ㆍ수입 품목의 허가 신청)
② (현행과 같음)
3. 제조판매 품목허가를 신청하는 품목을 그 품목과 동일한 품목에 대해 제조판매 품목허가를 받거나 품목신고를 한 자의 제조소에서 다음 각 목의 구분에 따라 제조하도록 위탁하는 경우: 제1항제6호에 해당하는 자료. 다만, 나목의 경우에는 1개 제조단위 이상의 실적 자료를 제출해야 한다.
가. 해당 제조판매 품목허가를 받거나 품목신고를 한 품목(일반의약품, 원료의약품 및 의약외품만 해당한다)과 모든 제조공정을 동일하게 제조하도록 하는 경우
나. 해당 제조판매 품목허가를 받거나 품목신고를 한 품목(전문의약품만 해당한다)과 제조공정, 제조설비, 제조단위, 의약품과 직접 접촉하는 용기ㆍ포장의 재질 및 종류를 모두 동일하게 제조하도록 하는 경우
※ 의약품 등의 안전에 관한 규칙 제4조제2항 변경대비표
? 의약품 제조 및 품질관리에 관한 규정(식약처 고시) 개정 및 시행
❍ (배경) 우리나라는 의약품실사상호협력기구(PIC/S) 가입국(‘14.7)으로서 의약품 제조 및 품질관리기준(GMP)의 국제조화 지속 추진중
❍ [별표 13] 적격성평가와 밸리데이션 시행(‘20.6.29)
- PIC/S 가이드라인 부속서(Annex 15)의 개정사항을 ‘의약품 제조 및 품질관리기준’ [별표 13] 적격성평가와 밸리데이션에 반영
※ [별표 13] 주요 개정사항
◈ QbD 적용 시 공정밸리데이션 방법을 선택 적용
◈ 국내외 실질적 운영현황 반영
- 적격성평가 및 밸리데이션에 외부자료 활용
- 순차적 실시하도록 규정한 적격성평가 및 밸리데이션의 동시 수행
◈ 국내외 실질적 운영현황 반영
- 위험평가에 따라 적격성평가 및 밸리데이션 범위·정도 등 결정
- 적격성평가 중요 단계 및 주요 권장기준 제시
- 운송검증 및 포장공정 밸리데이션, 세척밸리데이션 구체화 등
❍ [별표 17] 완제의약품 제조 개정(‘20.10.28 개정, ’21.10.29 시행)
- PIC/S GMP Part 1(완제의약품 GMP) 주요 개정사항을 ‘의약품 제조 및 품질관리기준’(식약처 고시) [별표 17]에 반영
※ [별표 17] 주요 개정사항
◈ GMP 기준에 의약품품질시스템(Pharmaceutical Quality System)의 도입
※ 의약품품질시스템(Pharmaceutical Quality System): 의약품 개발부터 생산 및 판매종료까지 지속적 개선을 촉진하는 관리 시스템으로 의약품 제조 및 품질관리기준과 품질위험관리를 포함
◈ 품질 결함에 대한 조사를 위해 유통 중인 제품을 수거하는 경우에는 이를 회수로 간주하지 않도록 개선
◈ 위탁자 및 수탁자 간 책임과 기준 등에 대한 의약품 제조 및 품질관리 기준의 명확화
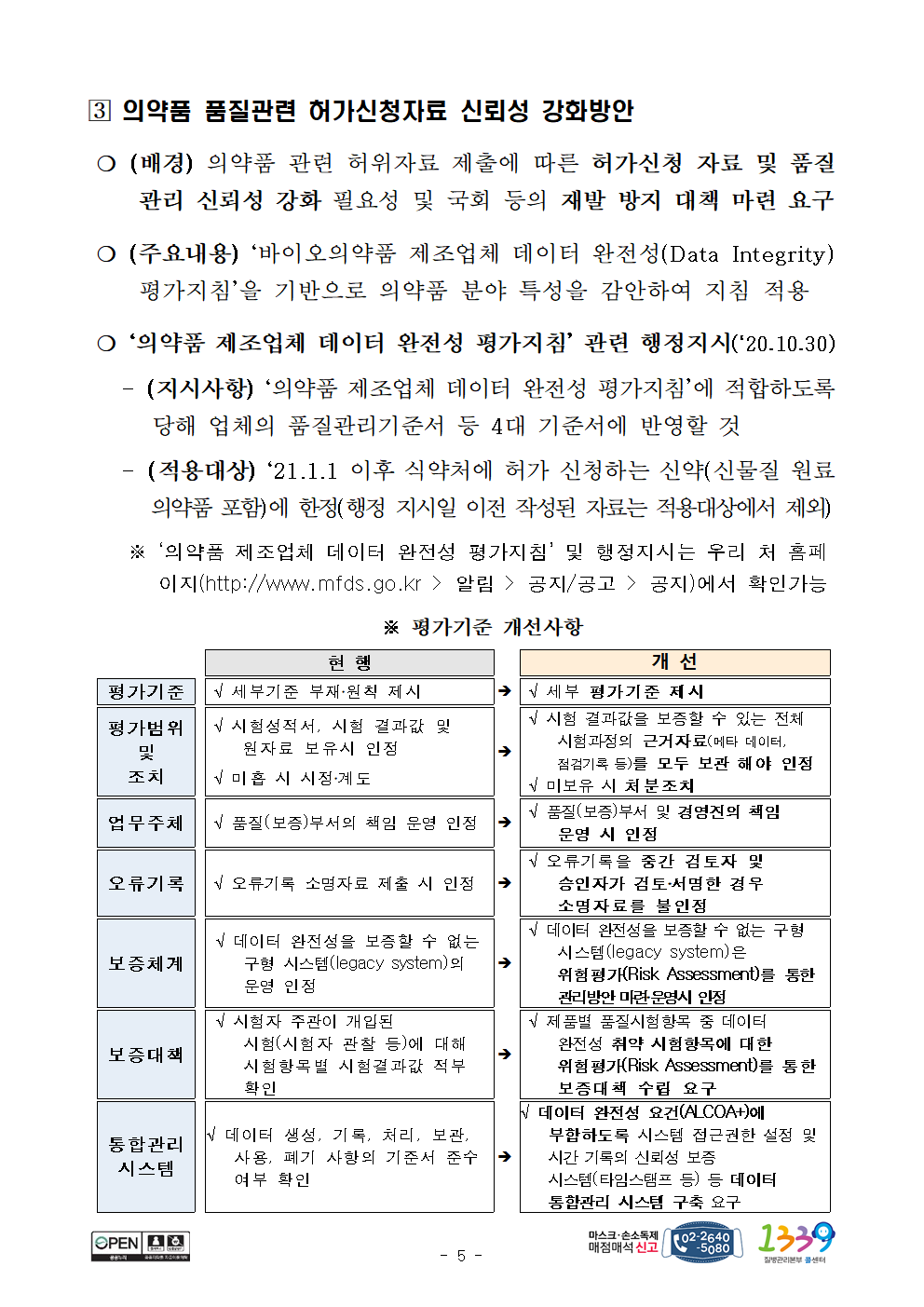
? 의약품 품질관련 허가신청자료 신뢰성 강화방안
❍ (배경) 의약품 관련 허위자료 제출에 따른 허가신청 자료 및 품질관리 신뢰성 강화 필요성 및 국회 등의 재발 방지 대책 마련 요구
❍ (주요내용) ‘바이오의약품 제조업체 데이터 완전성(Data Integrity) 평가지침’을 기반으로 의약품 분야 특성을 감안하여 지침 적용
❍ ‘의약품 제조업체 데이터 완전성 평가지침’ 관련 행정지시(‘20.10.30)
- (지시사항) ‘의약품 제조업체 데이터 완전성 평가지침’에 적합하도록 당해 업체의 품질관리기준서 등 4대 기준서에 반영할 것
- ( 적용대상) ‘21.1.1 이후 식약처에 허가 신청하는 신약(신물질 원료의약품 포함)에 한정(행정 지시일 이전 작성된 자료는 적용대상에서 제외)
※ ‘의약품 제조업체 데이터 완전성 평가지침’ 및 행정지시는 우리 처 홈페이지(http://www.mfds.go.kr > 알림 > 공지/공고 > 공지)에서 확인가능
현 행
개 선
평가기준
√ 세부기준 부재․원칙 제시
√ 세부 평가기준 제시
평가범위
및
조치
√ 시험성적서, 시험 결과값 및 원자료 보유시 인정
√ 미흡 시 시정․계도
√ 시험 결과값을 보증할 수 있는 전체 시험과정의 근거자료(메타 데이터, 점검기록 등)를 모두 보관 해야 인정
√ 미보유 시 처분조치
업무주체
√ 품질(보증)부서의 책임 운영 인정
√ 품질(보증)부서 및 경영진의 책임 운영 시 인정
오류기록
√ 오류기록 소명자료 제출 시 인정
√ 오류기록을 중간 검토자 및 승인자가 검토․서명한 경우 소명자료를 불인정
보증체계
√ 데이터 완전성을 보증할 수 없는 구형 시스템(legacy system)의 운영 인정
√ 데이터 완전성을 보증할 수 없는 구형 시스템(legacy system)은 위험평가(Risk Assessment)를 통한 관리방안 마련․운영시 인정
보증대책
√ 시험자 주관이 개입된 시험(시험자 관찰 등)에 대해 시험항목별 시험결과값 적부 확인
√ 제품별 품질시험항목 중 데이터 완전성 취약 시험항목에 대한 위험평가(Risk Assessment)를 통한 보증대책 수립 요구
통합관리
시스템
√ 데이터 생성, 기록, 처리, 보관, 사용, 폐기 사항의 기준서 준수 여부 확인
√ 데이터 완전성 요건(ALCOA+)에 부합하도록 시스템 접근권한 설정 및 시간 기록의 신뢰성 보증 시스템(타임스탬프 등) 등 데이터 통합관리 시스템 구축 요구
※ 평가기준 개선사항
? 코로나19 관련 사전 GMP 평가 개선방안
❍ (배경) ‘코로나 19’의 지속적 확산 및 장기화에 따라 수입의약품(원료의약품 포함) GMP 평가 개선 방안(1~3차)을 마련
❍ (시행사항) ‘코로나 19’ 상황 종료시까지 접수되는 허가 또는 등록 신청 건(기 접수 품목 및 변경 포함)에 대해 평가방안(1~3차 종합) 적용
< 평가방안 대비표 >
구분
종전
한시적 개편방안(종합, ‘20.10월~)
서류평가
DMF
11종 자료
(제조증명서로 갈음 가능)
좌동
★ 단, PIC/S 보고서 미보유 제조소 품목은 총리령 [별표 1의2에] 따른 자료 제출 필요(11종 자료)
품목허가
11종 자료
좌동
실태조사
(무균) 실태조사 실시다만, 3년 이내 식약처 실사결과 ‘적합’ 제조소는 생략
사전 실태조사 생략(전면 서류평가)
★ 다만, 종전지침*에 따른 실사대상 품목은 「약사법」 제69조의5에 따른 사후 실태조사 대상 우선 선정
* 의약품등 품목별 사전 GMP 평가 운영지침(‘16.12월)
(비무균) ▴5년 이내 식약처 실사결과 ‘적합’ 제조소 ▴(실사결과 ‘적합’ or PIC/S 가입국 소재) 제조소 중 PIC/S보고서 제출시 생략
(그 외 실태조사 실시)
* 전면 서류검토 진행에 따라 제조원 실태조사 시 확인을 사유로 GMP 평가 필수 자료(11종 자료 등) 미제출 불허용
'판교핫뉴스1' 카테고리의 다른 글
| 공공분야 최고의 드론조종사 가린다-2일 드론 교육훈련센터에서 드론 조종 경진대회 개최…40개 팀 참여 (0) | 2020.12.02 |
|---|---|
| '21년 신규 식품 영업자 대상 집합교육 한시적 유예-코로나19로 교육생 안전과 편의 위해 집합교육과 온라인교육 병행 운영 (0) | 2020.12.02 |
| 경제협력개발기구(OECD) 「경제전망」 발표[2020.12.01.]-‘20년 한국 성장률 전망 회원국 1위, 주요 20개국(G20) 2위 (0) | 2020.12.02 |
| ‘시스템반도체·바이오헬스·미래차’ 3대 신산업에 847억원 지원했다 (0) | 2020.12.02 |
| 2030년 부산세계박람회 유치의향 공식 표명-2030년 엑스포 유치 경쟁 본격 돌입 (0) | 2020.12.02 |